

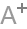
慢性肾脏病(CKD)已成为威胁人类健康的主要疾病之一。研究发现慢性肾衰竭患者存在肾小管周毛细血管(PTCs)减少,伴随血液灌流下降限制肾间质氧的供应,进而引起肾脏纤维化及肾小管萎缩,加速肾脏疾病进展。
另外糖尿病、高血压等CKD常见的继发性疾病中亦存在局部组织低氧现象,可见由于PTCs减少引起的组织低氧与器官纤维化密切相关。
近几年来,通过血管新生来改善肾小管间质低氧,延缓CKD疾病进展逐渐引起重视,但因新生血管易受炎症及血管通透性改变的影响而临床应用受限。本文着重讨论血管新生在改善肾组织低氧相关纤维化中的作用及其两面性,以及新型增加PTCs的临床干预措施。
一、肾间质低氧对肾脏纤维化作用
虽然全身有20%的血液流经肾脏,但其仍易遭受低氧损伤。在肾髓质区域,动脉与静脉紧密相连,脉管之间的气体弥散导致氧及营养的无效运输增加,造成肾髓质 低氧。哺乳动物中肾髓质氧分压为10 mmHg,肾髓质氧分压约为30 mmHg,远远低于如此高的肾血流所应有的氧分压。
另外近端肾小管及髓质升支粗段主要依靠Na/K⁃ATP酶进行高代谢活动,因此肾脏对低氧异常敏感,易受低氧损伤。一般情况下,血管内皮细胞处于静止状态,被壁细胞(周细胞、血管平滑肌细胞)包绕。
在低氧损伤时,内皮细胞去分化及再生能力受损,因此可在糖尿病肾小球硬化、慢性间质性肾炎、肾小球淀粉样变、失代偿性良性肾小球硬化、继发的恶性肾小球硬化等患者的肾脏病理中观察到PTCs 减少,且与血清肌酐呈负相关。
PTCs 减少引起氧及营养供应下降,导致肾组织低氧,低氧参与多种肾脏疾病进展,尤其是CKD。作者目前在C57BL/6衰老大鼠中的研究结果显示:衰老大鼠肾组织中出现局部低氧现象。
课题组前期研究结果显示在单侧输尿管梗阻(UUO)组动物模型中,26月龄衰老大鼠较3月龄年轻大鼠的肾小管间质纤维化面积大。
低氧促使肾小管上皮细胞凋亡并发生形态学变化,即上皮细胞间充质改变(epithelial⁃mesenchymal transition,EMT)。肾小管上皮细胞发生EMT,促使肾小管间质中细胞表面标志物E⁃钙黏素(E⁃cadherin)、增殖细胞核抗原 PCNA、α⁃平滑肌肌动蛋白(α⁃SMA)与细胞外基质Ⅰ型及Ⅲ型胶原的产生发生改变。
在衰老肾组织中,这些标志物的变化可能参与衰老相关的肾小管间质损伤与肾功能减退。另外自发的或因免疫性适应缺血状态可促进炎症细胞(巨噬细胞、T 淋巴细胞)的募集、加速炎症的进展,引起肾脏纤维化。
体内、外研究证明,低氧主要通过缺氧诱导因子(HIF)途径及非HIF 途径影响肾脏纤维化。在单肾切除、Thy⁃1肾炎、糖尿病大鼠、急性肾损伤(AKI)等模型中,HIF活化可延缓CKD。但亦有研究证明在肾小管上皮细胞 活化的HIF与CKD进展有关,其可促进肾脏纤维化。
因此不同类型肾脏疾病中HIF 作用可能不同。在肾脏疾病中,为维持PTCs,近端小管上皮细胞表达血管内皮生长因子(VEGF),而低氧通过刺激VEGF产生,参与肾脏纤维化。
二、传统促血管新生延缓肾脏疾病进展措施
研究发现,在人和动物CKD 疾病中,血管生成因子(VEGF、血管生成素⁃Tie系统)、内源性血管生成抑制因子(凝血酶敏感蛋白⁃1、血管抑素、内皮抑素等)表达失衡,导致毛细血管稀疏,加重肾脏低氧,促进肾脏纤维化,见图1。
因此传统的促血管新生措施主要从血管生成因子、血管抑制因子两方面来改善肾间质低氧,以达到延缓肾脏疾病进展目的。
1. 血管生成因子促进血管新生利与弊:
由壁细胞分泌的血管生成因子可促进血管新生及其重塑,其中以VEGF、血管生成素⁃Tie信号系统为主。VEGF的主要成分VEGF⁃A(由剪接变体亚 型、VEGF121、VEGF165、VEGF165b、VEGF189 及VEGF206 组成),通过结合VEGF 受体2(VEGFR⁃2/FLK⁃1)调节血管的生成。
血管生成素⁃Tie 信号系统在维持血管内皮静止及血管源信号通路传感性中起重要作用。血管生成素⁃1(Ang⁃1)在后肾间充质、分化成熟的肾小管上皮及足细胞中表达,血管 生成素⁃2(Ang⁃2)在直小血管附近的肾小管中表达,Ang⁃1、Ang⁃2 是Tie⁃2激动剂,二者互相竞争,Ang⁃1⁃Tie⁃2信号通路在维持内皮细胞稳定方面起重要作用。
在多种肾脏疾病中均可观察到VEGF⁃A表达下降,且与PTCs 减少有关。例如:应用3、12、24 月龄大鼠石蜡切片进行免疫组化染色、定量分析肾脏微血管变化及VEGF变化情况,结果显示24 月龄(衰老组)大鼠肾组织内肾小球和PTCs 明显减少,VEGF 减少,内皮细胞适应生存能力减退,出现血管生成障碍,因此利用血管生成因子诱导血管新生是必要的。
在抗Thy⁃1 肾炎、抗肾小球基底膜(GBM)抗体诱导的肾小球肾炎等模型中,使用VEGF治疗可促进肾小球内皮损伤的恢复,而使用VEGFR⁃1(VEGF受体)加速肾小球坏死、促使PTCs 减少及间质纤维化。
相比较野生型糖尿病鼠及VEGF⁃A 基因敲除鼠,糖尿病VEGF⁃A基因敲除鼠增加肾小球损伤、凋亡现象及蛋白尿。CKD患者中,可溶性血管内皮生长因子受体⁃1(sVEGFR⁃1)水平增加,其抑制VEGF表达,可导致内皮功能障碍,增加心血管发病率。
另外Futrakul 等对CKD 患者进行研究,发现Ang⁃2 表达上升,Ang⁃1 表达下降,Ang⁃1 可能在溶血性尿毒症综合征恢复过程中扮演重要角色,尤其是内皮细胞损伤,Ang⁃2 通过抑制Ang⁃1 与其共同受体Tie2结合,损伤内皮细胞,可作为评估CKD生存率的一个标志物。
事实上,血管生成因子促使血管新生改善肾间质低氧是存在争议的,原因在于VEGF 刺激产生的微脉管系统(MV)不成熟,弯曲、易渗漏,具有血管异质性。这导致新生MV渗出物增加,出现炎性反应。
另外组织间隙液压增加、毛细血管淤塞、氧及营养运输不平衡,缺少灌注刺激的脉管易于出现功能退化,局部灌注不足亦可引起低氧,二者协同导致肾组织缺血损伤。
另外血管生成素亦介导炎性反应及纤维化,如在大鼠缺血再灌注模型中,Ang⁃2/Ang⁃1比例失衡(Ang⁃2增加为主),伴随周细胞增殖,内皮细胞死亡,肾脏纤维化。在叶酸诱导的小鼠肾病模型中,即使PTCs未减少,Ang⁃1 亦可诱导炎性反应及纤维化。
2. 血管生成抑制因子抑制血管新生利与弊:
近十年来,血管生成抑制因子因其抑制血管生成的机制常被用来治疗肿瘤和其它的血管依赖性疾病。在PTCs密度减少的慢性肾脏疾病中,它的上调可加重肾组织低氧,但同时也可能抵抗血管新生所引发的炎症效应。
血管生成抑制因子,如凝血酶敏感蛋白(TSP),是一种多功能母细胞的糖蛋白,参与调节血管生成、伤口愈合、胶原纤维形成等过程,在肾脏疾病时高表达,是介导肾脏纤维化的重要介质。
体内、外研究证明,TSP 家族五位成员中,仅TSP1、TSP2 通过直接影响内皮细胞、间接影响生长因子的活化而抑制血管新生。在UUO 模型诱导的肾纤维化中,下调TSP⁃1可导致VEGF、PTCs密度增加,而间质细胞中的HIF⁃1α、III 型胶原及重组人结缔组织生长因子(CTGF)表达下降,因此抑制TSP⁃1 可通过增加PTCs密度改善肾脏纤维化。
其他血管生成抑制因子有:血管抑素、内皮抑素。血管抑素是纤维蛋白溶酶原的蛋白水解片段,可调节血管生成及炎性反应。Satoh 等认为血管生成障碍与衰老相关的肾脏MV减少有关,也是衰老相关肾脏病变的原因之一,而血管抑素可能是血管生成的潜在抑制剂。
他们检测不同月龄SD大鼠中血管抑素的表达,发现随着年龄增长血管抑素表达逐渐增加,认为可能与衰老相关的血管减少及间质损伤有关。内皮抑素是胶原ⅩⅧ的C⁃末端片段,是血管内皮生长因子的潜在调节者。
检测201 个CKD 患者及相同数目正常人血清中内皮抑素的表达,发现血清内皮抑素表达与CKD 密切相关。由此可见血管生成抑制因子可能通过降低PTCs密度,诱导肾间质低氧,加速疾病进展。
事实上,过表达的血管抑制因子在降低PTCs 密度的同时也具有抗炎作用。在链脲霉素诱导的糖尿病大鼠中,内源性血管抑素水平下降,提高血管抑素水平可改善蛋白尿及肾小球肥大,提示其可能具有抗炎、抗纤维化作用。
在单肾切除大鼠模型中,过表达血管抑素可改善肾间质纤维化及抑制巨噬细胞、T 淋巴细胞渗透,也提示血管抑制因子具有抗炎作用。在糖尿病肾病及其他CKD患者中,VEGF、血管生成素⁃Tie 系统等血管生成因子总体上表达下降,血管生成抑制因子如血管抑素、TSP1 等表达上调,正是这种不平衡,促进肾脏疾病进展。
而简单地通过单一的干预措施虽然可促进血管新生,增加肾小管周毛细血管网数目,有利于改善肾间质低氧,但亦可加重慢性炎症。因此,探索新型增加PTCs的干预措施势在必行。
3. 增加PTCs的新型干预措施:
单一的干预措施具有两面性,一方面促进血管新生,改善肾组织低氧,另一方面易导致组织炎症,见图2。目前,新型的干预措施从维持内皮细胞稳定、多血管因子级联反应、细胞疗法等方面增加PTCs,产生健康成熟的新生血管,改善肾间质低氧,延缓CKD进展。
内皮细胞活化可能促进肌成纤维细胞的活化或者向间质细胞的转变(EndoMT),造成PTCs 减少,加速疾病进展。类肝素硫酸蛋白聚糖(HSPGs)存在于内皮细胞表面可维持内皮细胞的抗凝及抗炎特性。
分泌神经素是一种血管原神经肽,在不同动物模型中,通过增加脉管密度改善组织灌注。体外实验研究显示分泌神经素主要依赖碱性成纤维细胞生长因子(bFGF)促进人类皮肤内皮细胞血管新生、增殖、凋亡,而硫酸类肝素作为辅因子促进这一过程。
硫酸葡聚糖是一种HSPGs类似物,在血栓性微血管病(TMA)模型中,阻止内皮细胞凝固及补体激活,改善肾小球内皮细胞损伤及PTCs稀疏。
多种血管因子的级联反应可能会改善单一血管因子刺激而引起的一系列问题。例如,在小鼠内皮细胞中脯氨酸羟化酶2(PHD⁃2)异质体的缺乏,使HIF稳定表达,可维持肿瘤血管的密度及大小稳定,易渗漏、弯曲、重塑的血管减少,成熟化血管增加。
目前对迁移性肾细胞癌治疗的细胞因子疗法中,靶向调控VEGFR、mTORC1 的因子不仅有效而且毒性小。然而由于产生抗药性,导致肿瘤化脉管系统重新产生,许多病人复发。因此多因子联合靶向调控多种血管生成信号通路,包括 VEGFR、PI3K 和mTORC1/2,可能成为治疗肾细胞癌的新型治疗方法。
内皮祖细胞(endothelial progenitor cells,EPCs)可把血管原细胞因子及生长因子运输到缺血器官,通过旁分泌逆转急性肾损伤。
研究证明,随着慢性疾病进展,血液循环中的内皮祖细胞下降。与正常人比较,CKD 患者EPCs低表达。与早期CKD(Ⅰ⁃Ⅲ)患者比较,晚期(Ⅳ⁃Ⅴ或者ESRD)患者EPCs 数目更低。血清VEGF 水平与循环中的EPCs水平密切相关。
在出现微量白蛋白尿的糖尿病患者中,循环内皮祖细胞数量下降,内皮细胞修复机制受损,可能导致EndoMT发生。在猪肾动脉狭窄的模型中,EPCs可减少内质网压力及氧化应激,增强肾脏生长因子表达,刺激新生血管的增殖与成熟,有利于微血管重塑,改善纤维化。
在经过缺血预处理的SD 大鼠缺血再灌组模型中,研究发现通过促进EPCs流动及募集,可改善肾脏损伤,且VEGF在此过程中可能扮演重要角色。另外,来源于EPCs的微泡亦可通过促进血管新生及阻止内皮细胞凋亡进而对急慢性损伤具保护作用。
在缺血再灌注损伤的大鼠模型中,静脉注射的微泡主要在PTCs 及肾小管细胞处聚集,通过促进肾小管细胞增殖、减少凋亡及白细胞浸润而保护肾脏。微泡可阻止PTCs 减少,减轻肾小球硬化及肾小管纤维化,预防慢性疾病对肾脏的损伤。
三、小结
CKD在全球范围内发病率逐年增加,研究预防及延缓肾脏疾病进展的方法逐渐引起重视。PTCs减少是慢性肾脏疾病的特征之一,它引起肾间质低氧,促使肾小 管上皮细胞、内皮细胞等向肌成纤维细胞(间质纤维化的关键效应细胞)转化,以及血管生成因子与抑制因子表达失衡,引起肾脏纤维化,对CKD 进展具有重要意义。
因此研究血管新生在CKD中的作用至关重要。由于传统的促血管新生方案具有两面性,新型增加PTCs 干预措施可能有助于解决这一难题,但仍需进一步的验证。



